Genentech issued the following announcement on Oct. 30.
Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), announced today that the U.S. Food and Drug Administration (FDA) has accepted the company’s Biologics License Application (BLA) for satralizumab for the treatment of adults and adolescents with neuromyelitis optica spectrum disorder (NMOSD). The European Medicines Agency (EMA) has also validated the company’s Marketing Authorization Application (MAA) for satralizumab, granting it Accelerated Assessment. The FDA decision and the EMA’s Committee for Medicinal Products for Human Use (CHMP) recommendation are expected in 2020.
“People living with NMOSD experience unpredictable relapses that can cause permanent neurological damage, and although there have been significant strides recently in understanding the disease, more approved options are needed with different treatment approaches. Satralizumab has shown robust efficacy sustained for 96 weeks and significantly reduced the risk of relapse across a broad patient population, while offering self-administered subcutaneous dosing every four weeks,” said Levi Garraway, M.D., Ph.D., chief medical officer and head of Global Product Development. “The FDA and EMA’s acceptances of the satralizumab applications bring us one step closer to providing a new medicine to thousands of people impacted by NMOSD, and we are working with the health authorities to make satralizumab available as soon as possible.”
The satralizumab applications are based on positive results from two Phase III studies, SAkuraStar and SAkuraSky, evaluating the efficacy and safety of satralizumab as a monotherapy and in combination with baseline immunosuppressant therapy, respectively. In the SAkuraStar study, satralizumab monotherapy achieved a 55% reduction in the risk of relapses compared to placebo in the overall study population of aquaporin-4 antibody (AQP4-IgG) seropositive and seronegative patients (Hazard Ratio [HR]=0.45, 95% Confidence Interval [CI]: 0.23-0.89; p=0.0184). Satralizumab achieved a 74% reduction (HR=0.26, 95% CI: 0.11-0.63; p=0.0014) in the larger (~67%) subgroup of AQP4-IgG seropositive patients, who tend to experience a more severe disease course. In the overall satralizumab-treated population, 76.1% were relapse-free at 48 weeks, and 72.1% relapse-free at 96 weeks, compared to 61.9% and 51.2% with placebo, respectively. Data from the AQP4-IgG seropositive subgroup showed that 82.9% were relapse-free at 48 weeks and 76.5% relapse-free at 96 weeks when treated with satralizumab, compared to 55.4% and 41.1% with placebo, respectively.
Additionally, the SAkuraSky study data showed a 62% reduction in the risk of relapses compared to placebo in the overall study population (HR=0.38, 95% CI: 0.16-0.88; p=0.0184) when used in combination with baseline immunosuppressant therapy, and a 79% reduction in the risk of relapses in AQP4-IgG seropositive patients (HR=0.21, 95% CI: 0.06-0.75; p=0.0086). In the overall satralizumab-treated population, 88.9% were relapse-free at 48 weeks, and 77.6% were relapse-free at 96 weeks, compared to 66.0% and 58.7% with placebo, respectively. Data from the AQP4-IgG seropositive subgroup showed that 91.5% were relapse-free at 48 and 96 weeks when treated with satralizumab, compared to 59.9% and 53.3% with placebo, respectively.
Overall, the proportion of patients with serious adverse events was similar between the satralizumab and placebo treatment groups in both studies. A lower rate of infections (including serious infections) was observed in patients treated with satralizumab compared with the placebo group. Most adverse events were mild to moderate, and the most common adverse events in the satralizumab group were urinary tract infection and upper respiratory tract infection in the SAkuraStar study and upper respiratory tract infection, nasopharyngitis (common cold) and headache in the SAkuraSky study.
The FDA previously granted Breakthrough Therapy Designation to satralizumab for the treatment of NMOSD in December 2018.
About neuromyelitis optica spectrum disorder (NMOSD)
NMOSD is a rare, lifelong and debilitating autoimmune disease of the central nervous system that primarily damages the optic nerve(s) and spinal cord, causing blindness, muscle weakness and paralysis. People with NMOSD experience unpredictable, severe relapses directly causing cumulative, permanent, neurological damage and disability. In some cases, relapse can result in death. NMOSD affects over 10,000 people in Europe, 15,000 people in the United States and up to hundreds of thousands of people worldwide. The disease is most common among non-Caucasian women in their 30s and 40s.
NMOSD is commonly associated with pathogenic antibodies (AQP4-IgG) that target and damage a specific cell type, called astrocytes, resulting in inflammatory lesions of the optic nerve(s), spinal cord and brain. AQP4-IgG antibodies are detectable in the blood serum of around two-thirds of NMOSD patients.
Although most cases of NMOSD can be confirmed through a diagnostic test, people living with the condition are still frequently misdiagnosed with multiple sclerosis. This is due to overlapping characteristics of the two disorders, including a higher prevalence in women, similar symptoms and the fact that both are relapse-based conditions.
About satralizumab
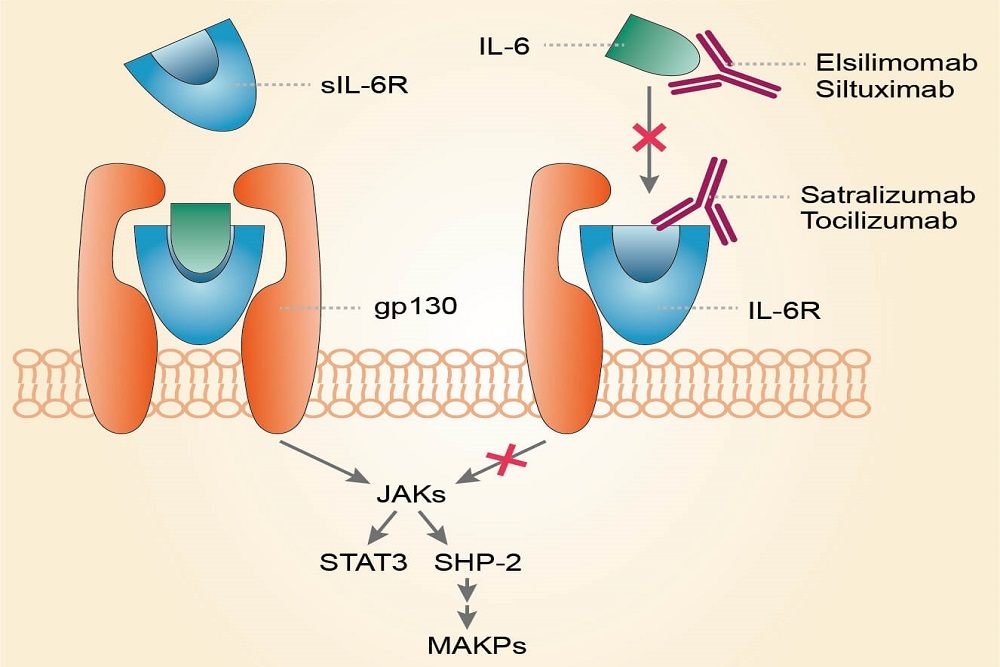
Satralizumab is an investigational humanized monoclonal antibody that targets the interleukin-6 (IL-6) receptor. The cytokine IL-6 is thought to be a key driver of NMOSD, triggering the inflammation cascade and leading to damage and disability. Positive Phase III results for satralizumab, as both monotherapy and in combination with baseline immunosuppressant therapy, suggest that IL-6 inhibition may be an effective therapeutic approach for NMOSD. The Phase III clinical development program for satralizumab includes two studies: SAkuraStar and SAkuraSky.
About SAkuraStar and SAkuraSky in NMOSD
SAkuraStar is a Phase III multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of satralizumab monotherapy administered to patients with NMOSD. The primary endpoint is the time to first protocol-defined relapse (PDR), adjudicated by an independent review committee in the double-blind period.
Ninety-five patients 20-70 years of age were randomized to either of the following two treatment groups in a 2:1 ratio: satralizumab (120 mg) or placebo. Both treatments were administered subcutaneously at Week 0, 2, and 4. The subsequent treatment was continued at 4-week intervals. The double-blind treatment period ended when the total number of PDRs had reached 44 or at 1.5 years after the enrollment of the last patient, whichever occurred first. After experiencing a PDR or upon completion of the study, patients in both groups were offered treatment with satralizumab in an open-label extension period. Patients with AQP4-IgG seropositive or seronegative neuromyelitis optica (NMO, as defined by the diagnostic criteria in 2006) and those with AQP4-IgG seropositive NMOSD were enrolled.
SAkuraSky is a Phase III multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of satralizumab added to baseline immunosuppressant therapy in patients with NMOSD. The primary endpoint was the time to first relapse as adjudicated by an independent review committee in the double-blind period.
Eighty-three male and female patients 13-73 years of age were randomized to either of the following two treatment groups in a 1:1 ratio: satralizumab (120 mg) or placebo added to baseline immunosuppressant therapy (azathioprine, mycophenolate mofetil and/or corticosteroids). Both treatments were administered subcutaneously at Week 0, 2, and 4. The subsequent treatment was continued at 4-week intervals. The double-blind treatment ended when patients experienced a PDR; the study ended when the total number of PDRs reached 26. After experiencing a PDR or upon completion of the study, patients in both groups were offered treatment with satralizumab in an open-label extension period. Patients with AQP4-IgG seropositive or seronegative neuromyelitis optica (NMO, as defined by diagnostic criteria in 2006) and those with AQP4-IgG seropositive NMOSD were enrolled.
About Genentech in neuroscience
Neuroscience is a major focus of research and development at Genentech and Roche. The company’s goal is to develop treatment options based on the biology of the nervous system to help improve the lives of people with chronic and potentially devastating diseases. Genentech and Roche have more than a dozen investigational medicines in clinical development for diseases that include multiple sclerosis, spinal muscular atrophy, neuromyelitis optica spectrum disorder, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, Duchenne muscular dystrophy and autism.
About Genentech
Founded more than 40 years ago, Genentech is a leading biotechnology company that discovers, develops, manufactures and commercializes medicines to treat patients with serious and life-threatening medical conditions. The company, a member of the Roche Group, has headquarters in South San Francisco, California. For additional information about the company, please visit http://www.gene.com.
Original source can be found here.




